New Paper - "Fission Gas in Thoria"
Our recent computational work using density functional theory on “Fission gas in thoria” has been accepted and is now available from the Journal of Nuclear Materials.
- Navaratnarajah Kuganathan, Partha S. Ghosh, Conor OT. Galvin, Ashok K. Arya, Bijon K. Dutta, Gautam K. Dey, Robin W. Grimes, “Fission gas in thoria”, J. Nucl. Mater., 485 (2017) 47–55. doi:10.1016/j.jnucmat.2016.12.011
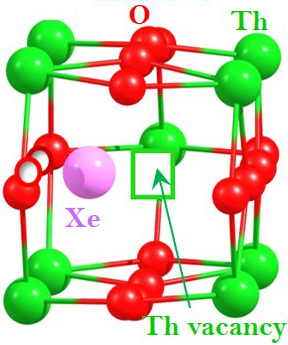
This study uses first-principles density functional theory together with a dispersion correction (DFT+D) to calculate the energetics of neutral and charged point defects and defect cluster geometries in ThO2. It also investigates their interaction with Xe and Kr. ThO2 has been identified as a possible alternative nuclear fuel, partly because spent ThO2 fuels give rise to considerably smaller inventories of minor actinides, especially Pu. Furthermore, ThO2 is a highly stable oxide, and exhibits higher thermal conductivity, higher melting temperature, higher corrosion resistance and lower thermal expansion compared to UO2. At high fuel temperatures, fission gas atoms migrate and are accommodated at point defect sites in the fuel matrix. Over time some of these aggregate into bubbles. Formation of bubbles is important as it leads to swelling and degrades mechanical properties of the material. In order to understand fuel performance, it is necessary to understand the interaction of gas atoms with point defects.